
The introduction of biophysical methods into molecular dynamics for the study of protein-protein interactions
Molecular interactions play a key role in all the branch of analytical sciences. Among these interactions, those involving proteins are particularly fascinating as they comprise a large number of degree of freedom and are present in all biological processes. Understanding how proteins move and interact with neighboring molecules is necessary for solving critical puzzles in molecular biology and design potent new inhibitors. Until now, these interactions can be modeled by means of basic docking but these techniques usually badly represent protein flexibility and solvent effects.
No single technique can at present span the whole range of typical time and length scales relevant for a protein biological function and interaction. Nevertheless, biophysical methods like NMR, SAXs or SANS can provide valuable informations in terms of dynamics. By using NMR spin relaxation, residual dipolar couplings or the attachment of paramagnetic probes combined with molecular dynamics, it is possible to restrict the conformational space explored by large molecular assemblies and extract valuable informations. The movie represents the use of quaternions in the determination of the rotational diffusion tensor in complexes or globular proteins.
Protein-Protein interactions - Ubiquitin and polyubiquitin chains
Among the existing post-translational modifications, ubiquitination is probably the most versatile and intriguing modification. It occurs through the covalent attachment of ubiquitin to target proteins. Ubiquitination of proteins directs the modified proteins to different cellular fates such as translocation, assembly or degradation, depending on the type of tagging. Ubiquitin Binding Domains, which are modular elements, are devoted to the recognition of the ubiquitin tag and direct ubiquitinated proteins to the right pathway. Our group focuses on the lysosomal degradation and more particularly on the interaction between Ubiquitin Binding Domains (UBDs) and ubiquitin or polyubiquitin chains. It is of prime importance to characterize these interactions to better understand the underlying biological events in lysosomal degradation ( Example of the VHS/Ubiquitine complex solved by NMR). Our research projects are carried out by means of NMR (Nuclear Magnetic Resonance) as well as molecular docking. The primary goal is to characterize the structural and dynamical properties of the studied complexes involved in lysosomal degradation.
Linkage Dependence of Polyubiquitin Conformations
The question of how different polyubiquitin signals are recognized is central to understanding the specificity of various types of polyubiquitination. Thus, K48-linked chains, a universal signal for proteasomal degradation, under physiological conditions adopt a closed conformation where functionally important residues L8, I44, and V70 are sequestered at the interface between two adjacent Ub monomers. By contrast, K63-linked chains, which act as a nonproteolytic regulatory signal, adopt an extended conformation that lacks hydrophobic interubiquitin contact. Little is known about the functional roles of the so-called “noncanonical” chains (linked via K6, K11, K27, K29, or K33, or linked head-to-tail), and no structural information on these chains is available, except for information on the crystal structure of the head-to-taillinked diubiquitin (Ub2). Our results show that the eight possible Ub2 chains can be divided into two groups: chains linked via K6, K11, K27, or K48 are predicted to form a closed conformation, whereas chains linked via K29, K33, or K63, or linked head-to-tail are unable to form such a contact due to steric occlusion.

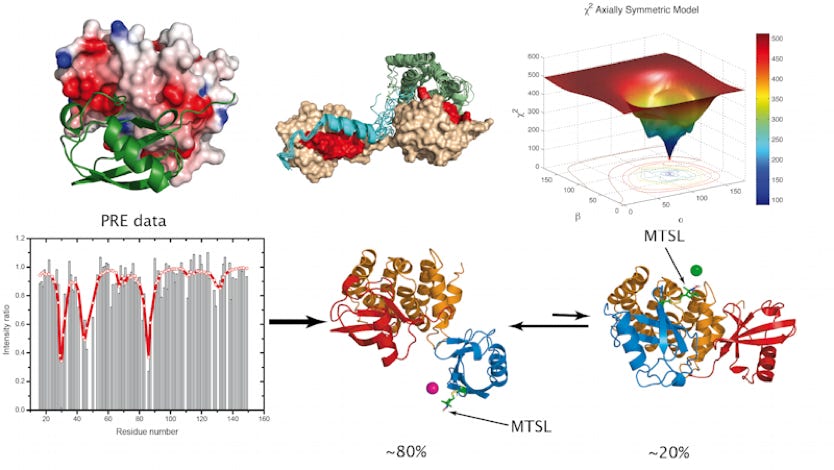
Determining domain orientation in multi-domain proteins
The basic idea of determining domain orientation in multi-domain proteins is based on the orientational dependence of some NMR parameters (i.e. by extracting the rotational diffusion or alignment tensor). First, the orientation of the Principal Axes Frame (PAF) of the tensor of interest is determined for each individual domain using relaxation (diffusion tensor) or RDCs (alignment tensor) measurements for the whole molecule. This orientation is generally characterized by three Euler angles that define the PAF with respect to the atom coordinate (PDB) frame for a given domain. Since the domains reorient together as a single entity, these individual PAFs represent properties of the whole molecule, experienced by each individual domain. Therefore, as a second step, these principal axes frames (PAFs) determined for the individual domains are aligned such that their corresponding axes become parallel to each other.
